Ligand-based virtual screening benchmarks (LBVS) are notoriously tricky to interpret: hidden biases, flattering evaluations for deep learning models, and plain memorization all get in the way. While the issues with DUD-E [dude] or MUV [muv] are well documented in literature [dude_bias] , the last ‘standing’ benchmark was specifically designed to avoid this problem:
LIT-PCBA [litpcba] benchmark includes 15 targets, with 7,844 confirmed active compounds and 407,381 confirmed inactive ones, closely replicating real-world experimental screening decks in terms of hit rate and potency distribution while tackling the issue of biases present in other ligand-based virtual screening (LBVS) benchmarks using AVE Unbiasing procedure, [ave] preventing the potential “memorization” on the benchmarked ligands. [cheese]
The authors of Data Leakage and Redundancy in the LIT-PCBA Benchmark [litpcba_audit] are aiming to probe if this benchmark is robust enough to score novel methods fairly, or if it suffers from similar kind of issues, notably from redundancies and train-test leakages affecting the extent to which it really evaluates generalization.^benchmark_splits
The LIT-PCBA Audit paper discussed in this blog has two versions. I was writing main parts of this blog and creating a github fork before the version 2 was released. The main results (and conclusions) of these two versions do not substantially differ, but the latest version answers to common objections and dedicates a whole section on why stereoisometry was omited: mainly as a diagnostic step to expose leakage which is causing memorization, not as a modeling recommendation. For a charitable reading, please skip to section “Can stereoisomers cause memorization?” of this blog.
There is ‘leakage’… but it’s a stereochemical collapse
The jupyter notebook in the LIT-PCBA Audit paper which runs the leakage and redundancy checks prints out summary of the findings: [litpcba_audit_repo]
================================================================================
OVERALL OUTRAGEOUS STATS ACROSS ALL RECEPTORS
--------------------------------------------------------------------------------
Number of query molecules also in training set (overall): 2
Number of query molecules also in validation set (overall): 1
Number of inactive molecules in both train and val (overall): 2491
Number of repeating molecules in queries (overall): 5
Number of repeating molecules in active_T (overall): 15
Number of repeating molecules in inactive_T (overall): 2945
Number of repeating molecules in active_V (overall): 1
Number of repeating molecules in inactive_V (overall): 789
================================================================================
Before deduplication, the authors canonicalized the representations of the molecules in the benchmark using rdkit with explicit flag isomericSmiles=False, removing stereoisomeric information from the original SMILES strings in the process.
For consistency, stereochemistry was removed, as is standard practice in ligand-based modeling where 2D fingerprints are often used [litpcba_audit]
I was able to reproduce this [litpcba_audit_fork] and discovered that from the 2,945+15 supposedly duplicate or leaked molecules in the training set and 789+1 in the validation set, all of them are stereoisomers: while their non-isomeric smiles representation is identical, the original isomeric smiles provided in the benchmark are different. Running the same code with when I switched to isomericSmiles=True (and ignored the query set smiles yields ^query_ligands ):
================================================================================
OVERALL OUTRAGEOUS STATS ACROSS ALL RECEPTORS
--------------------------------------------------------------------------------
================================================================================
With stereo preserved, identity leakage (exact isomeric-SMILES matches across splits) drops to zero.

Can stereoisomers cause memorization?
It seemed to me as a silly mistake: the authors found an “egregious data leakage”, which turns out to be consisting entirely from stereoisomers ignored during canonicalization. Isn’t it then a bit uncharitable to label the whole benchmark as “fundamentally compromised” when just stereoisomers are found? Shall we strip stereoisomers away from benchmarks to prevent a train-test “leakage”? Do they test memorization, not generalization? Or is this just some misconception?
Since the second version was released,[litpcba_audit_v2] clarifying the intention to stress-test the splits for “stereo-agnostic identity leakage”, I started to take this question seriously. Let’s have a look on some examples I picked from the ALDH1 target splits in LIT-PCBA:
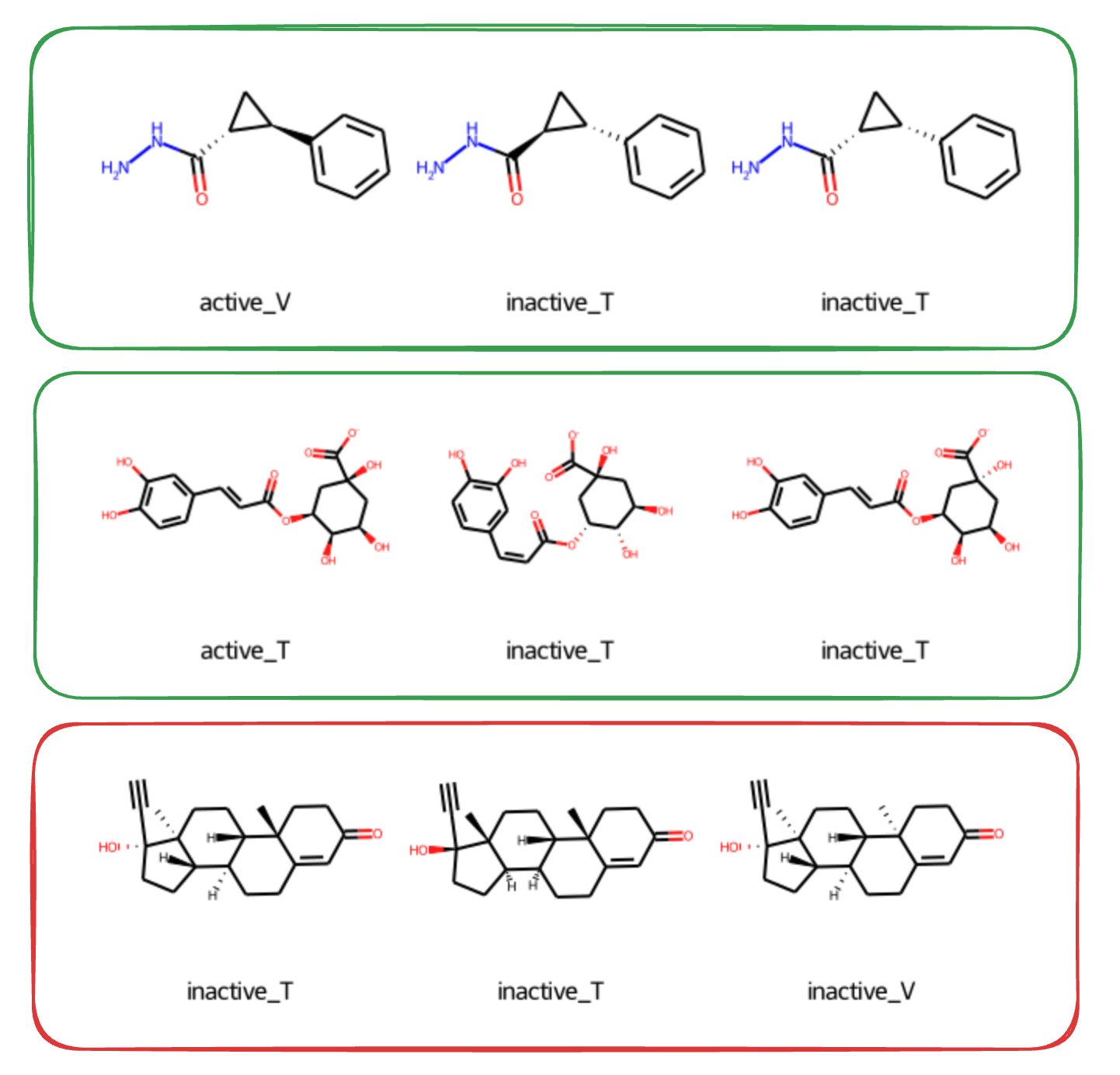
- Green: stereoisomers have different labels: across train (T) and test (V) split some of them are active, some are inactive
- Red: stereoisomers have the same label (all of them are inactive or all active)
In the green (conflicting-label) cases, it can be very hard or effectively impossible to predict for 2D methods. Especially when there is only one stereoisomer in the training set, for instance labelled as inactive, and then others in the test set labelled differently, say as active. Vice versa: if the labels are the same across splits, a method which isn’t sensitive to stereochemistry might get 100% accuracy on these pairs of stereoisomers. And on the others it will get close to 0%.
Furthermore, without treating stereoisomers with a special care, there will be likely many more cases with the same label and just a fraction of those with conflicting labels in a way which realiably tests generalization. Most often, we will find a group of stereoisomers where all of them are inactive. This supports the worry about memorization: a method ignoring stereochemistry will memorize all of the positive pairs (or negative pairs) and the reported enrichment factor may be artificially inflated:
stereochemistry is scarcely rewarded at evaluation: there is exactly one instance of label-discordant stereoisomers in the validation set. Meanwhile, stereoisomers recur across splits almost exclusively with the same label [litpcba_audit_v2]
2D and 3D methods
So far I have taken for granted the assumption that stereoisomers are problematic, “indistinguishable” so to say for most of existing methods. But this is not universally true. Even with the “2D fingerprints” used by the authors of the audit you can choose to preserve stereochemistry if you want. Here is an example:
from rdkit import Chem
from rdkit.Chem import AllChem, DataStructs
# Example molecules: one pair of enantiomers to show stereochemical sensitivity
smiles_a = "F[C@H](Cl)Br" # R-config
smiles_b = "F[C@@H](Cl)Br" # S-config
mols = [Chem.MolFromSmiles(s) for s in (smiles_a, smiles_b)]
# ECFP4 parameters
radius = 2 # ECFP4 = radius 2;
n_bits = 4096 # The length author's use
fps_stereo = [AllChem.GetMorganFingerprintAsBitVect(m, radius=radius,
nBits=n_bits,
useChirality=True) # ⬅️ crucial
for m in mols]
fps = [AllChem.GetMorganFingerprintAsBitVect(m, radius=radius,
nBits=n_bits,
useChirality=False)
for m in mols]
# Compare the two fingerprints:
tanimoto_stereo = DataStructs.TanimotoSimilarity(fps_stereo[0], fps_stereo[1])
tanimoto = DataStructs.TanimotoSimilarity(fps[0], fps[1])
print(f"Tanimoto similarity (if chirality matters): {tanimoto_stereo:.3f}")
print(f"Tanimoto similarity: {tanimoto:.3f}")
Tanimoto similarity (if chirality matters): 0.778
Tanimoto similarity: 1.000
Moreover, the paper is focusing mainly on 3D methods such as CHEESE [cheese] , Surflex [surflex] or [ROCS] . Since all of these are sensitive to stereochemistry, the question whether their performance is inflated because of memorization is not straightforward to answer: these methods are expected to handle stereochemistry and in “3D” a large fraction of the stereoisomers will look very different in their shape. 3D methods are avoiding this sort of “representational collapse” by design and their proneness to these pitfalls shall reflect their actual stereochemical sensitivity.
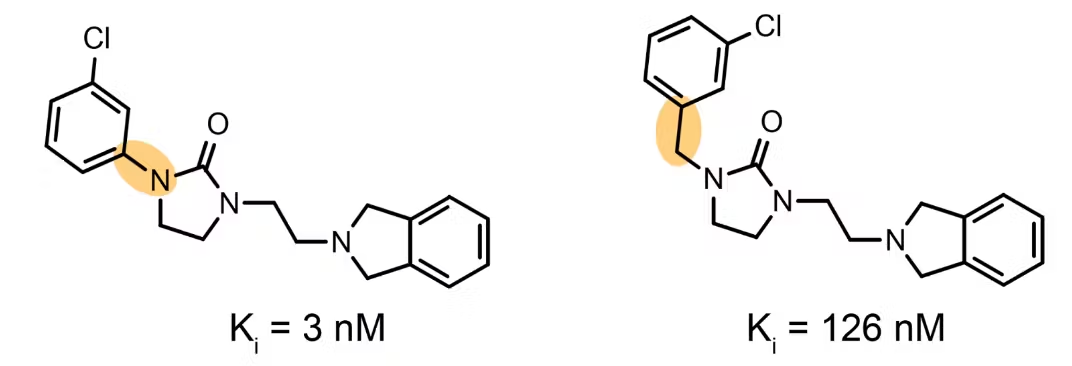
Stereoisomers: “activity cliffs” of LBVS benchmarks?
A textbook activity cliff is a matched molecular pair that differs by only a single atom or functional group, yet shows a dramatic jump in bioactivity. These pairs are infamously hard for machine-learning and QSAR models: their 2-D fingerprints (and hence their Tanimoto similarity) are essentially 1.0, so the input looks almost identical to the algorithm—while the output (the activity) is starkly different.[molecule_ace] Stereoisomer pairs with opposing labels pose exactly the same challenge, in fact some of them would be classified as activity cliffs too.
Is the benchmark compromised?
The “egregious leakage” in the LIT-PCBA Audit largely comes from collapsing stereochemistry during canonicalization. However, this doesn’t mean the audit is pointless; it highlights an important diagnostic: when the number of stereo-consistent and stereo-inconsistent clusters is imbalanced like it is in LIT-PCBA, stereo insensitive (or “2D biased”) pipelines can reward memorization on the compounds with consistent labels, while underestimating failures on the few truly discordant pairs.
My take:
- Stereochemistry needs to be included where possible and benchmarked too, not ignored during canonicalization.
- Make “stereo-cliffs’ explicit. Annotate stereoisomer clusters and report stratified metrics (consistent vs. discordant).
- Publish the stereo-sensitivity gap. Pair every method with a chirality-off variant to surface memorization.
With those tweaks, every LBVS bechmark containing such “2D-leakage” remains a useful and realistic. Not “fundamentally compromised,” but clearer about where models generalize and where they merely remember.
References
Footnotes
In LIT-PCBA there are five files with compounds provided for each target. Train set SMILES (both active and inactive compounds): [“active_T.smi”, “inactive_T.smi”]. Test set SMILES (both active and inactive compounds): [“active_V.smi”, “inactive_V.smi“]. And ligands in mol2 format (active).
Important note: the ‘query ligands’ in mol2 files are not used in building machine learning models where you learn on the training set and predict on the test set. As clarified by the benchmark authors they “were considered just to benchmark the target sets and remove target classes for which simple 2D and 3D similarity searches let to null enrichments.”